- IONIQUES (STRUCTURES)
- IONIQUES (STRUCTURES)Si la présence d’ions au sein de solutions salines ou de sels fondus semblait depuis longtemps bien établie, notamment par des études de conductivité électrique, l’extension de la notion d’ion à l’édifice cristallin reposait par contre sur un postulat dont le succès résidait essentiellement dans l’obtention par le calcul, pour les énergies réticulaires de certains types de cristaux, de valeurs très satisfaisantes. Les développements des techniques de rayons X, en offrant la possibilité de dresser pour un cristal de véritables cartes de densités électroniques, ont permis de démontrer d’une manière directe l’existence d’ions au sein de certains types de réseaux, et de valider ainsi le postulat de la liaison ionique.Un cristal ionique est formé par un assemblage géométrique simple d’ions chargés positivement, les cations, et d’ions chargés négativement, les anions, l’énergie qui assure la cohésion du cristal, ou l’énergie réticulaire, étant d’origine essentiellement électrostatique.Le nombre des composés à caractère ionique prédominant est très important; cette catégorie englobe une grande partie des composés de la chimie minérale et des constituants de nombreuses variétés de roches. Cependant, les cristaux purement ioniques sont rares, ils se limitent pratiquement aux fluorures et chlorures des métaux alcalins lourds; en effet, une évolution vers un caractère plus covalent – ou homopolaire – apparaît rapidement pour les autres composés.Rayons ioniquesW. L. Bragg en 1920, puis V. M. Goldschmidt en 1926, étudiant de manière systématique un grand nombre de structures ioniques, montraient que les ions pouvaient être considérés comme des sphères électriquement chargées possédant des rayons bien caractéristiques et constants; en particulier, dans la série des halogénures alcalins, les variations régulières des distances internucléaires cation-anion, obtenues avec précision à partir de leurs paramètres cristallins déterminés par diffraction X (cf. CRISTAUX Cristallographie), étaient en bon accord avec cette hypothèse. Ces valeurs ne permettaient cependant pas à elles seules la connaissance des divers rayons ioniques; la détermination préalable du rayon d’un ion particulier s’imposait donc.A. Landé supposait alors que, dans la structure de l’iodure de lithium correspondant au plus petit cation Li+ et au plus gros anion I-, les sphères anioniques étaient en contact; il en déduisait immédiatement le rayon de l’ion I-, qui, dans ce cas, conditionnait seul l’arête de la maille du cristal de LiI. Dès lors, de proche en proche, l’ensemble des rayons ioniques pouvait être déterminé.D’autres méthodes de partage des distances internucléaires furent ensuite proposées, en particulier par Goldschmidt; elles conduisirent à des résultats très voisins de ceux de Landé. Le tableau ci-après rassemble les valeurs des rayons de quelques ions.L. Pauling, quelques années plus tard, proposa une méthode de calcul plus théorique fondée sur l’influence de la charge nucléaire effective Zeff sur la distribution des électrons périphériques d’un ions. D’autres physiciens ont proposé, depuis, d’autres méthodes plus élaborées de calcul direct; les résultats ne diffèrent pas sensiblement de ceux de Goldschmidt ou de Landé.La possibilité de déterminer la densité électronique en tout point d’un cristal permet actuellement de tracer une véritable carte de cette densité électronique, comme celle qui est représentée sur la figure 1; l’intégration des diverses courbes isoélectroniques permet alors de connaître le nombre total d’électrons environnant chaque noyau; dans le cas du chlorure de sodium, pris ici comme exemple, le transfert de l’électron périphérique 3 s du sodium sur l’atome de chlore est total (Na+: 11 漣 1 = 10 électrons, Cl-: 17 + 1 = 18 électrons). Ce résultat expérimental confirme donc pleinement la théorie semi-empirique de l’édifice ionique. De plus, l’examen des densités électroniques le long d’un segment joignant les deux noyaux de sodium et de chlore permet d’observer un minimum très net correspondant à la limite entre les deux sphères anionique et cationique; les rayons ioniques obtenus ainsi directement (RNa+ = 0,118 nm, RCl- = 0,164 nm) diffèrent cependant très notablement des valeurs précédemment admises; ces divergences montrent bien les limites actuelles du concept de rayon ionique.Énergie de liaison dans les cristaux ioniquesLes cristaux ioniques sont généralement caractérisés par des forces de cohésion importantes; il en résulte, pour ces composés, des températures de fusion relativement élevées. Au sein d’une même série, telle que les fluorures alcalins, elles diminuent quasi linéairement lorsque la distance internucléaire cation-anion augmente; il en va donc de même pour l’énergie du réseau, ou énergie réticulaire.M. Born et J. R. Mayer ont montré que l’énergie d’un cristal ionique pouvait se diviser en deux termes: une énergie d’interaction coulombienne entre les divers ions porteurs de charges électriques, dont la résultante négative H1, diminue lorsque l’on déplace ces ions de l’infini pour assembler le cristal (fig. 2); une énergie de répulsion positive H2, qui apparaît et augmente brutalement lorsque les nuages électroniques des divers ions commencent à se superposer. La résultante de ces deux termes H possède un minimum correspondant à la position d’équilibre D entre cations et anions. La valeur de l’énergie réticulaire H est alors donnée par l’expression:
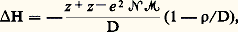 où z + et z - représentent respectivement le nombre des charges du cation et de l’anion, e la charge électrique de l’électron, 類 le nombre d’Avogadro, 福 une constante reliée au coefficient de compressibilité du cristal et 紐 la constante de Madelung, grandeur qui ne dépend que de la géométrie du cristal. Cette relation rend parfaitement compte de l’énergie réticulaire d’un cristal purement ionique, énergie que l’on peut d’ailleurs déterminer indirectement par des méthodes thermodynamiques (cycle de Born-Haber par exemple); elle explique également la diminution de la stabilité du réseau des fluorures alcalins avec l’augmentation de la distance internucléaire D.Coordinence dans les cristaux ioniquesD’une manière générale et pour des structures simples, le réseau d’un composé ionique est déterminé par la coordinence des ions – c’est-à-dire par le nombre d’ions de signe opposé immédiatement à leur contact – et, plus spécialement, par la coordinence de l’ion le plus petit, le cation. L’énergie d’interaction coulombienne augmente avec la coordinence, mais celle-ci ne peut dépasser une limite imposée par les dimensions respectives de l’anion et du cation: lorsque l’on place un nombre croissant de sphères anioniques identiques autour d’un cation donné, au-delà d’un certain nombre limite, les anions ne sont plus en contact avec le cation central, trop petit, qui «flotte» littéralement dans sa cavité; à partir de cet instant, la distance internucléaire cation-anion (D) devenant supérieure à la somme des rayons cationique et anionique, l’énergie réticulaire du réseau ainsi formé diminuerait conformément à la relation de Born et Mayer. On en déduit les valeurs limites suivantes pour le rapport x du rayon du cation au rayon de l’anion pour diverses coordinences:– coordinence 8 cubique: x 麗 0,732;– coordinence 6 octaédrique: x 麗 0,414;– coordinence 4 tétraédrique: x 麗 0,225;– coordinence 3 triangulaire: x 麗 0,155.On citera pour exemple trois composés: le chlorure de césium CSCl, le chlorure de sodium NaCl et le sulfure de zinc ZnS (blende), pour lesquels le rapport x prend successivement les valeurs 0,92, 0,54 et 0,37, et dont les structures sont décrites à l’article CRISTAUX - Cristallographie.Polarisation dans les cristaux ioniquesDans les cristaux ioniques, le phénomène de polarisation correspond à une déformation du nuage électronique des ions primitivement supposés parfaitement sphériques sous l’influence du champ électrique des ions environnants. Ce phénomène, qui tend à donner à la liaison ionique un caractère directionnel, constitue un premier pas vers la liaison covalente de nature essentiellement dirigée; cette évolution est d’autant plus marquée qu’il existe, dans le réseau, de gros anions déformables – on dit encore polarisables – et des cations très petits et fortement chargés susceptibles de produire un champ électrique intense. La diminution de la coordinence d’un cation avec sa taille s’accompagne donc d’une évolution progressive du caractère ionique au caractère covalent de la liaison. On citera comme exemple d’une telle évolution les trois structures des oxydes de zirconium Zr2 (variété de haute température), de titane TiO2 (rutile) et de silicium Si2 (cristobalite) représentées à la figure 3. Si l’oxyde Zr2 peut être considéré à juste titre comme un oxyde typiquement ionique, doué même à l’état solide d’une conductivité électrique de type ionique relativement importante, la silice Si2 correspond déjà à un oxyde assez nettement covalent, étant donné le pouvoir polarisant du silicium, porteur de quatre charges électriques formelles et caractérisé par un très faible rayon «ionique» 0,039 nm environ.Évolution du caractère ionique au caractère moléculaire d’un cristalUn cristal moléculaire peut être défini comme formé par l’assemblage de molécules distinctes dont la cohésion est assurée par des forces du type Van der Waals. Celles-ci, relativement peu intenses, ne confèrent à ce type de cristaux qu’une faible stabilité; les cristaux moléculaires ont donc des températures de fusion ou de sublimation très basses. L’apparition d’un caractère moléculaire sera décelable dans un cristal dès lors que se formeront des groupements atomiques électriquement neutres, plus ou moins volumineux, isolés les uns des autres, donc faiblement liés entre eux.Une telle évolution se traduit, dans un premier stade, par la formation de structures à feuillets, véritables molécules géantes bidimensionnelles reliées entre elles par des forces de Van der Waals ; ces cristaux sont facilement clivables (le sulfure de molybdène MoS2 par exemple). On peut observer, dans le second stade, la formation de chaînes isolées. Le terme ultime de cette évolution correspond au groupement quasi ponctuel qu’est la molécule.L’apparition plus ou moins marquée de ce caractère moléculaire est généralement liée à la présence dans le réseau de très gros anions fortement polarisables. Les diverses structures des halogénures Mo3, CrCl3 AlBr3 sont un exemple de cette évolution. Le réseau, tridimensionnel dans le cas du fluorure de molybdène Mo3, dont l’anion -, relativement petit, est peu polarisable, devient bidimensionnel dans le cas du chlorure de chrome trivalent CrCl3, dont l’anion Cl- est déja beaucoup plus polarisable (cf. tableau); le réseau de CrCl3 est formé par l’empilement de feuillets constitués d’ions Cr3+ entre deux couches d’ion Cl- (fig. 4). Le réseau du bromure d’aluminium AlBr3, dont l’anion Br- est encore plus volumineux, correspond cette fois à un empilement de molécules dimérisées Al2Br6 constituées de deux tétraèdres (AlBr4) présentant une arête commune. On notera l’évolution simultanée de la coordinence cationique, qui passe de six dans le réseau du fluorure à quatre dans celui du bromure.Il apparaît ainsi que les cristaux purement ioniques ne constituent qu’une gamme très étroite de matériaux et qu’une classification en cristaux ioniques, covalents, moléculaires et métalliques semble beaucoup trop tranchée; entre ces divers cas limites, tous les intermédiaires sont en effet possibles. La théorie du réseau ionique permet cependant d’apporter de nombreux et fructueux renseignements dans le cas de beaucoup d’entre eux.
où z + et z - représentent respectivement le nombre des charges du cation et de l’anion, e la charge électrique de l’électron, 類 le nombre d’Avogadro, 福 une constante reliée au coefficient de compressibilité du cristal et 紐 la constante de Madelung, grandeur qui ne dépend que de la géométrie du cristal. Cette relation rend parfaitement compte de l’énergie réticulaire d’un cristal purement ionique, énergie que l’on peut d’ailleurs déterminer indirectement par des méthodes thermodynamiques (cycle de Born-Haber par exemple); elle explique également la diminution de la stabilité du réseau des fluorures alcalins avec l’augmentation de la distance internucléaire D.Coordinence dans les cristaux ioniquesD’une manière générale et pour des structures simples, le réseau d’un composé ionique est déterminé par la coordinence des ions – c’est-à-dire par le nombre d’ions de signe opposé immédiatement à leur contact – et, plus spécialement, par la coordinence de l’ion le plus petit, le cation. L’énergie d’interaction coulombienne augmente avec la coordinence, mais celle-ci ne peut dépasser une limite imposée par les dimensions respectives de l’anion et du cation: lorsque l’on place un nombre croissant de sphères anioniques identiques autour d’un cation donné, au-delà d’un certain nombre limite, les anions ne sont plus en contact avec le cation central, trop petit, qui «flotte» littéralement dans sa cavité; à partir de cet instant, la distance internucléaire cation-anion (D) devenant supérieure à la somme des rayons cationique et anionique, l’énergie réticulaire du réseau ainsi formé diminuerait conformément à la relation de Born et Mayer. On en déduit les valeurs limites suivantes pour le rapport x du rayon du cation au rayon de l’anion pour diverses coordinences:– coordinence 8 cubique: x 麗 0,732;– coordinence 6 octaédrique: x 麗 0,414;– coordinence 4 tétraédrique: x 麗 0,225;– coordinence 3 triangulaire: x 麗 0,155.On citera pour exemple trois composés: le chlorure de césium CSCl, le chlorure de sodium NaCl et le sulfure de zinc ZnS (blende), pour lesquels le rapport x prend successivement les valeurs 0,92, 0,54 et 0,37, et dont les structures sont décrites à l’article CRISTAUX - Cristallographie.Polarisation dans les cristaux ioniquesDans les cristaux ioniques, le phénomène de polarisation correspond à une déformation du nuage électronique des ions primitivement supposés parfaitement sphériques sous l’influence du champ électrique des ions environnants. Ce phénomène, qui tend à donner à la liaison ionique un caractère directionnel, constitue un premier pas vers la liaison covalente de nature essentiellement dirigée; cette évolution est d’autant plus marquée qu’il existe, dans le réseau, de gros anions déformables – on dit encore polarisables – et des cations très petits et fortement chargés susceptibles de produire un champ électrique intense. La diminution de la coordinence d’un cation avec sa taille s’accompagne donc d’une évolution progressive du caractère ionique au caractère covalent de la liaison. On citera comme exemple d’une telle évolution les trois structures des oxydes de zirconium Zr2 (variété de haute température), de titane TiO2 (rutile) et de silicium Si2 (cristobalite) représentées à la figure 3. Si l’oxyde Zr2 peut être considéré à juste titre comme un oxyde typiquement ionique, doué même à l’état solide d’une conductivité électrique de type ionique relativement importante, la silice Si2 correspond déjà à un oxyde assez nettement covalent, étant donné le pouvoir polarisant du silicium, porteur de quatre charges électriques formelles et caractérisé par un très faible rayon «ionique» 0,039 nm environ.Évolution du caractère ionique au caractère moléculaire d’un cristalUn cristal moléculaire peut être défini comme formé par l’assemblage de molécules distinctes dont la cohésion est assurée par des forces du type Van der Waals. Celles-ci, relativement peu intenses, ne confèrent à ce type de cristaux qu’une faible stabilité; les cristaux moléculaires ont donc des températures de fusion ou de sublimation très basses. L’apparition d’un caractère moléculaire sera décelable dans un cristal dès lors que se formeront des groupements atomiques électriquement neutres, plus ou moins volumineux, isolés les uns des autres, donc faiblement liés entre eux.Une telle évolution se traduit, dans un premier stade, par la formation de structures à feuillets, véritables molécules géantes bidimensionnelles reliées entre elles par des forces de Van der Waals ; ces cristaux sont facilement clivables (le sulfure de molybdène MoS2 par exemple). On peut observer, dans le second stade, la formation de chaînes isolées. Le terme ultime de cette évolution correspond au groupement quasi ponctuel qu’est la molécule.L’apparition plus ou moins marquée de ce caractère moléculaire est généralement liée à la présence dans le réseau de très gros anions fortement polarisables. Les diverses structures des halogénures Mo3, CrCl3 AlBr3 sont un exemple de cette évolution. Le réseau, tridimensionnel dans le cas du fluorure de molybdène Mo3, dont l’anion -, relativement petit, est peu polarisable, devient bidimensionnel dans le cas du chlorure de chrome trivalent CrCl3, dont l’anion Cl- est déja beaucoup plus polarisable (cf. tableau); le réseau de CrCl3 est formé par l’empilement de feuillets constitués d’ions Cr3+ entre deux couches d’ion Cl- (fig. 4). Le réseau du bromure d’aluminium AlBr3, dont l’anion Br- est encore plus volumineux, correspond cette fois à un empilement de molécules dimérisées Al2Br6 constituées de deux tétraèdres (AlBr4) présentant une arête commune. On notera l’évolution simultanée de la coordinence cationique, qui passe de six dans le réseau du fluorure à quatre dans celui du bromure.Il apparaît ainsi que les cristaux purement ioniques ne constituent qu’une gamme très étroite de matériaux et qu’une classification en cristaux ioniques, covalents, moléculaires et métalliques semble beaucoup trop tranchée; entre ces divers cas limites, tous les intermédiaires sont en effet possibles. La théorie du réseau ionique permet cependant d’apporter de nombreux et fructueux renseignements dans le cas de beaucoup d’entre eux.
Encyclopédie Universelle. 2012.